
In recent years, next-generation DNA sequencers have produced an increasingly detailed picture of how genes are expressed at the molecular level. The transcriptional output of these genes - the RNA copies produced from DNA - has revealed a richness of complexity in transcript structure and function, providing insights into the molecular-level properties of cancers and other diseases.
One of the most powerful methods for analyzing RNA transcripts is the Cap Analysis of Gene Expression (CAGE) protocol developed at the RIKEN OSC. A unique approach, CAGE enables not only high-throughput gene expression profiling, but also simultaneous identification of transcriptional start sites (TSS) specific to each tissue, cell or condition.
With HeliScopeCAGE, the OSC research team has adapted the existing CAGE protocol for use with the revolutionary HeliScopeTM Single Molecule Sequencer. Unlike earlier sequencers, the HeliScope Sequencer does not employ polymerase chain reaction (PCR) amplification to multiply a small number of DNA strands for analysis, a process which can introduce biases into data. Instead, the HeliScope Sequencer actually sequences the DNA strand itself, enabling direct, high-precision measurement.
In a paper published in Genome Research, RIKEN researchers confirm that this direct approach reduces biases and generates highly reproducible data from between 5 micrograms to as little as 100 nanograms of total RNA. A comparison using a leukemia cell line (THP-1) and a human cervical cancer cell line (HeLa) further shows that results from the technique are closely correlated to those from traditional microarray analysis. By making possible high-precision gene expression analysis from tiny samples, HeliScopeCAGE greatly expands the scope of research at the OSC, strengthening the institute's role in Japan as a hub for next-generation genome analysis.
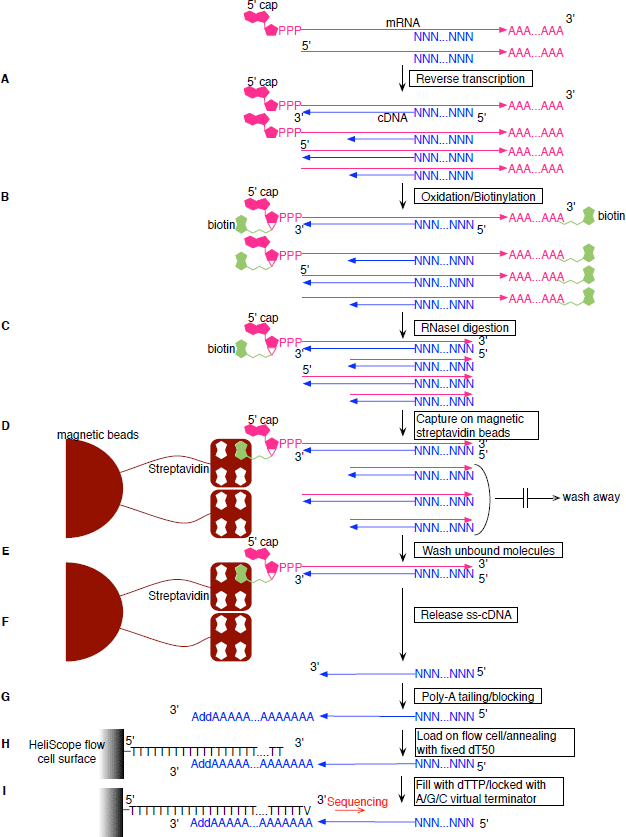
Figure 1: HeliScopeCAGE protocol workflow. (A) Reverse transcription. cDNA is synthesized using SuperScript III and random N15 primer. (B) Oxidation/Biotinylation. The cap structure is oxidized with sodium peroxide and biotinylated with biotin (long arm) hydrazine. (C) RNase I digestion. Single strand RNA is digested with RNase I. (D) Capture on magnetic streptavidin beads. Biotinylated RNA/cDNA hybrid molecules are captured using magnetic streptavidin beads. (E) Wash unbound molecules. Unbound RNA/DNA hybrid molecules are washed away. (F) Release ss-cDNA. Captured RNA/DNA hybrid molecules are treated with RNase H and RNase I, then heat treated. (G) Poly-A tailing/blocking. Released cDNA is poly-A tailed using terminal deoxynucleotidyl transferase and dATP, then blocked with biotin-ddATP. (H) Load on flow cell. Blocked poly-A tailed cDNA is loaded on the HeliScope flow cell channel and anneals with the dT 50 surface. (I) Fill with dTTP/locked with A/G/C virtual terminator. After annealing of cDNA, the single strand poly-A tail part is filled with DNA polymerase, dTTP, and an A/G/C virtual terminator which is used for the HeliScope sequencing to lock the poly-T termini. The library is then ready for sequencing.
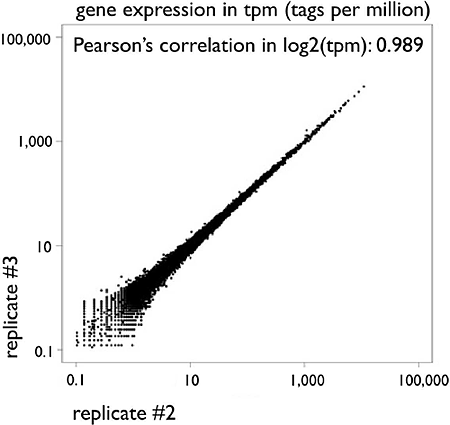
Figure 2: HeliScopeCAGE is a highly quantitative reproducible technology: Scatter plot of gene expressions between two technical replicates of HeliScopeCAGE on THP-1 RNA (5ug total RNA as starting material). The CAGE tag counts mapped within +/- 500bp from RefSeq transcription starting site are normalized as TPM (tags per million) with the library sizes.
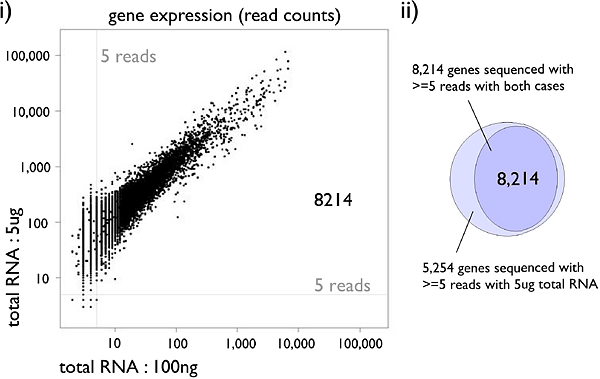
Figure 3: HeliScopeCAGE is a highly quantitative reproducible technology: (i) Gene expressions between different starting materials, 5ug and 100ng total RNA of THP-1. Scatter plots of the two profiles with read counts. (ii) The number of detected genes with each profile. A gene is considered detected when 5 or more reads are obtained.
References
- Mutsumi Kanamori-Katayama, Masayoshi Itoh, Hideya Kawaji, Timo Lassmann, Shintaro Katayama, Miki Kojima, Nicolas Bertin, Ai Kaiho, Noriko Ninomiya, Carsten O. Daub, Piero Carninci, Alistair R. R. Forrest and Yoshihide Hayashizaki. Unamplified Cap Analysis of Gene Expression on a single molecule sequencer. Genome Research (2011). doi: 10.1101/gr.115469.110