
技術の研究
CAGEの基本は、マウスエンサイクロペディアプロジェクトで開発した一連の完全長cDNA技術です。
CAGEライブラリを作成するには、はじめに、細胞や組織から抽出した全RNAから、ランダムプライマーまたはオリゴdTプライマーを用いて相補鎖cDNAを合成し、キャップ・トラッピング法によりcDNAの5’末端を選別します。ついで、RNasIを用いてRNA鎖を除去して得られる一本鎖cDNAの5’末端にビオチン化したリンカーを結合します。このリンカーには、クローニングに必要な認識部位、目印になる短い特異的塩基配列およびエンドヌクレアーゼ認識部位(MmeI)が含まれています。第2鎖cDNAを合成した後、クラスII制限酵素(MmeI)で5’末端から20塩基を切り出します。これをタグ配列といいます。切り出されたタグ配列の3’側に二本のリンカーを結合させます。このリンカーは別の制限酵素認識部位(XmajI)を含んでいます。ストレプトアビジンコートした磁気ビーズを用いて、ビオチン化したcDNAタグのみを取り出して精製します。ついで、両端のリンカーにある認識部位を制限酵素で切断し、リンカーの間に挟まれたタグを切り出します。このようにして得られたCAGEタグのDNA配列を決定し、ゲノム上にマップしていきます。
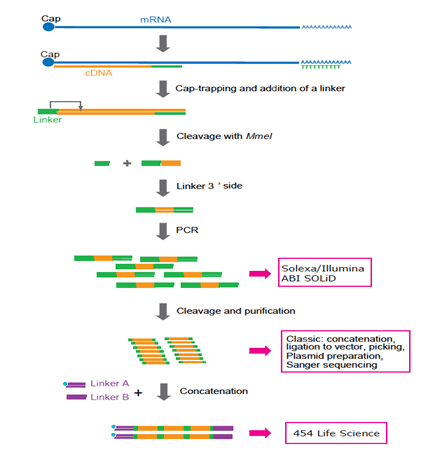
当初、CAGEタグは10個以上を連鎖結合(コンカテマー化)して、シーケンシングを行いました。FANTOM3では、これによって、転写開始点のゲノムワイドなマップが得らました。
さらに、最近のショットガン型シーケンサーの導入により、CAGE法は、全く新しいフェーズに入りました。
CAGEは定量的な遺伝子発現解析の強力なツールとなったのです。これは、重複したCAGEタグをすべてシーケンシングしてゲノムにマップし、マップされたタグの数によって各遺伝子の発現量を計測できるようになったためです。
ショットガンシーケンサーでは、1runあたり1,000,000タグ~10,000,000タグをシーケンシングすることができるので、CAGE法に適用すると、1コピーRNA/1細胞~10細胞の頻度のRNA分子を、理論的には99.9%以上の確率で捕らえることができます。
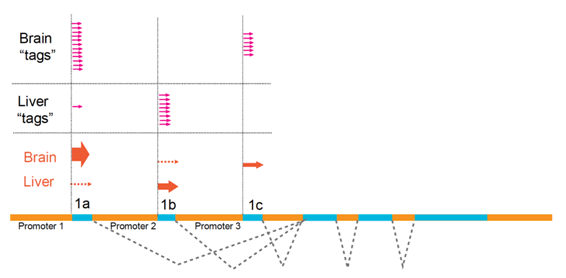
現在、プロモーターごとの遺伝子発現をゲノムワイドに調べる方法は、CAGEしかありません。
技術の応用
新しい診断と治療法の開発のための、CAGEの利用拡大を目指しています。CAGEは疾患モデルの特徴解明や新しいバイオマーカーおよび腫瘍マーカーの同定にも応用されています。たとえば、がん細胞と正常細胞の発現プロファイルを比較することにより、がん特異的に活性化しているプロモーターを特定することができるのです。
参考文献
- T. Shiraki, S. Kondo, S. Katayama, K. Waki, T. Kasukawa, H. Kawaji, R. Kodzius, A. Watahiki, M. Nakamura, T. Arakawa, S. Fukuda, D. Sasaki, A. Podhajska, M. Harbers, J. Kawai, P. Carninci, and Y. Hayashizaki, Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage, Proc Natl Acad Sci U S A,100, 15776-15781 (2003)
- R. Kodzius, M. Kojima, H. Nishiyori, M. Nakamura, S. Fukuda, M. Tagami, D. Sasaki, K. Imamura, C. Kai, M. Harbers, Y. Hayashizaki, and P. Carninci, CAGE: cap analysis of gene expression, Nat Methods,3, 211-222 (2006)